



RT-qPCR dikembangkan dari teknologi PCR biasa.Ini menambahkan bahan kimia fluoresen (pewarna fluoresen atau probe fluoresen) ke sistem reaksi PCR tradisional, dan mendeteksi proses anil dan perpanjangan PCR secara real time sesuai dengan mekanisme pendaran yang berbeda.Perubahan sinyal fluoresen pada medium digunakan untuk menghitung jumlah perubahan produk pada setiap siklus PCR.Saat ini, metode yang paling umum adalah metode pewarna fluoresen dan metode probe.
Metode pewarna neon:
Beberapa pewarna fluoresen, seperti SYBR Green Ⅰ, PicoGreen, BEBO, dll., tidak memancarkan cahayanya sendiri, tetapi memancarkan fluoresensi setelah berikatan dengan alur minor dsDNA.Oleh karena itu, pada awal reaksi PCR, mesin tidak dapat mendeteksi sinyal fluoresen.Ketika reaksi berlanjut ke annealing-extension (metode dua langkah) atau tahap ekstensi (metode tiga langkah), untai ganda dibuka saat ini, dan polimerase DNA baru. Selama sintesis untai, molekul fluoresen digabungkan dalam alur minor dsDNA dan memancarkan fluoresensi.Ketika jumlah siklus PCR meningkat, semakin banyak pewarna bergabung dengan dsDNA, dan sinyal fluoresen juga terus ditingkatkan.Ambil SYBR Green Ⅰ sebagai contoh.
Metode penyelidikan:
Probe Taqman adalah probe hidrolisis yang paling umum digunakan.Ada kelompok fluoresen di ujung probe 5′, biasanya FAM.Probe itu sendiri adalah urutan yang melengkapi gen target.Ada kelompok quenching fluoresen di ujung 3′ fluorofor.Menurut prinsip transfer energi resonansi fluoresensi (Transfer energi resonansi Förster, FRET), ketika kelompok fluoresen reporter (molekul fluoresen donor) dan kelompok fluoresensi quenching (molekul fluoresen akseptor) Ketika spektrum eksitasi tumpang tindih dan jaraknya sangat dekat (7-10nm), eksitasi molekul donor dapat menginduksi fluoresensi molekul akseptor, sementara autofluoresensi melemah.Oleh karena itu, pada awal reaksi PCR, ketika probe bebas dan utuh dalam sistem, gugus fluoresen pelapor tidak akan memancarkan fluoresensi.Saat anil, primer dan probe mengikat ke template.Selama tahap ekstensi, polimerase terus mensintesis rantai baru.DNA polimerase memiliki aktivitas eksonuklease 5′-3′.Saat mencapai probe, DNA polimerase akan menghidrolisis probe dari template, memisahkan kelompok fluoresen reporter dari kelompok fluoresen quencher, dan melepaskan sinyal fluoresen.Karena ada hubungan satu-ke-satu antara probe dan template, metode probe lebih unggul daripada metode pewarna dalam hal akurasi dan sensitivitas pengujian.
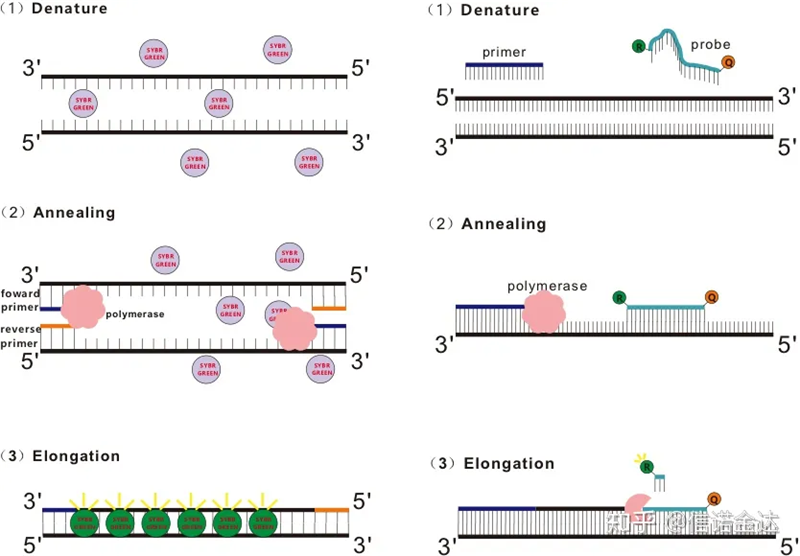
Gambar 1 Prinsip qRT-PCR
Desain primer
Prinsip:
Primer harus dirancang di daerah konservasi seri asam nukleat dan memiliki spesifisitas.
Yang terbaik adalah menggunakan urutan cDNA, dan urutan mRNA juga dapat diterima.Jika tidak, cari tahu desain wilayah cds dari urutan DNA.
Panjang produk kuantitatif fluoresen adalah 80-150bp, terpanjang adalah 300bp, panjang primer umumnya antara 17-25 basis, dan perbedaan antara primer hulu dan hilir tidak boleh terlalu besar.
Konten G+C antara 40% dan 60%, dan 45-55% adalah yang terbaik.
Nilai TM antara 58-62 derajat.
Cobalah untuk menghindari dimer primer dan self-dimer, (tidak muncul lebih dari 4 pasang basa komplementer berturut-turut) struktur hairpin, jika tidak dapat dihindari, buat ΔG<4.5kJ/mol* Jika Anda tidak dapat memastikan bahwa gDNA telah dihapus selama transkripsi balik Bersihkan, yang terbaik adalah merancang primer dari ujung intron *3′ tidak dapat dimodifikasi, dan untuk menghindari daerah kaya AT, GC, hindari T/C, A/G continuous structure (2-3) primer dan non-
Spesifik Homologi urutan yang diamplifikasi secara heterogen lebih disukai kurang dari 70% atau memiliki 8 homologi basa komplementer.
Basis data:
Pencarian CottonFGD dengan kata kunci
Desain primer:
Desain primer IDT-qPCR
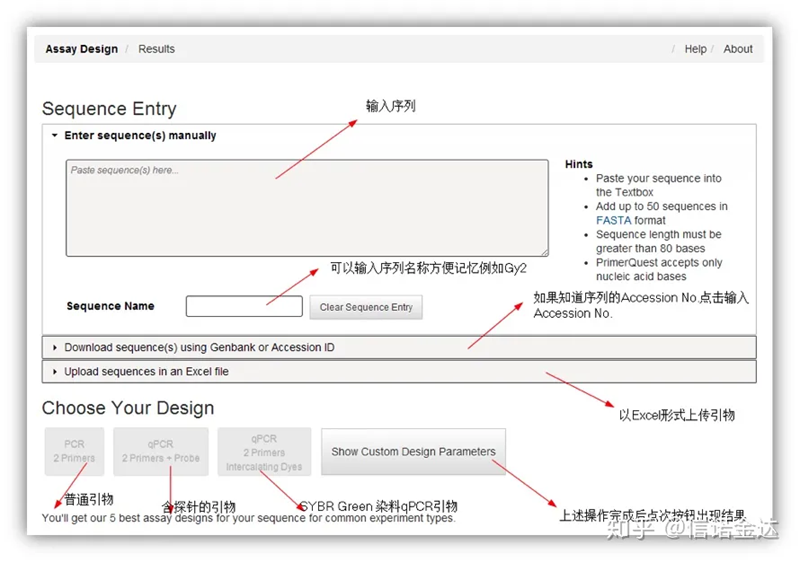
Fig2 halaman alat desain primer online IDT
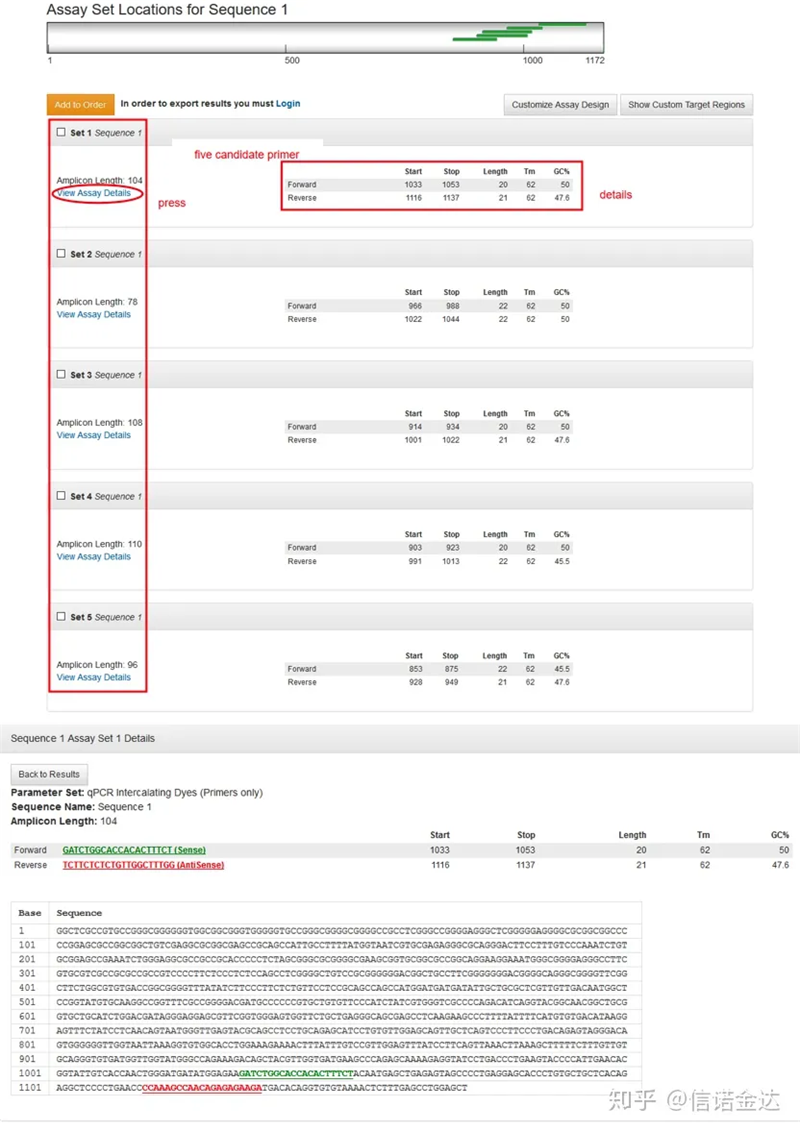
Tampilan halaman hasil Fig3
Desain primer lncRNA:
lncRNA:langkah yang sama dengan mRNA.
miRNA:Prinsip metode stem-loop: Karena semua miRNA adalah sekuens pendek sekitar 23 nt, deteksi PCR langsung tidak dapat dilakukan, sehingga alat sekuens stem-loop digunakan.Urutan batang-loop adalah DNA beruntai tunggal sekitar 50 nt, yang dapat membentuk struktur jepit rambut dengan sendirinya.3 'Ujung dapat dirancang sebagai urutan yang melengkapi fragmen parsial miRNA, kemudian miRNA target dapat dihubungkan ke urutan batang-loop selama transkripsi terbalik, dan panjang total dapat mencapai 70bp, yang sejalan dengan panjang dari produk yang diamplifikasi ditentukan oleh qPCR.Tailing desain primer miRNA .
Deteksi khusus amplifikasi:
Basis data ledakan online: ledakan CottonFGD berdasarkan kesamaan urutan
Ledakan lokal: Lihat menggunakan Blast+ untuk melakukan ledakan lokal, linux dan macos dapat langsung membuat database lokal, sistem win10 juga dapat dilakukan setelah menginstal ubuntu bash.Buat basis data ledakan lokal dan ledakan lokal;buka ubuntu bash di win10.
Perhatikan: Kapas dataran tinggi dan kapas pulau laut adalah tanaman tetraploid, sehingga hasil ledakan seringkali berupa dua atau lebih kecocokan.Di masa lalu, menggunakan CD NAU sebagai basis data untuk melakukan ledakan cenderung menemukan dua gen homolog dengan hanya sedikit perbedaan SNP.Biasanya, dua gen homolog tidak dapat dipisahkan dengan desain primer, sehingga diperlakukan sama.Jika terdapat indel yang jelas, primer biasanya dirancang pada indel, tetapi hal ini dapat menyebabkan struktur sekunder primer. Energi bebas menjadi lebih tinggi, menyebabkan penurunan efisiensi amplifikasi, tetapi hal ini tidak dapat dihindari.
Deteksi struktur sekunder primer:
Langkah:buka oligo 7 → masukkan urutan template → tutup sub-jendela → simpan → temukan primer pada template, tekan ctrl+D untuk mengatur panjang primer → analisis berbagai struktur sekunder, seperti badan dimerisasi sendiri, heterodimer, jepit rambut, ketidaksesuaian, dll. Dua gambar terakhir pada Gambar 4 adalah hasil pengujian primer.Hasil dari primer depan bagus, tidak ada struktur dimer dan jepit rambut yang jelas, tidak ada basa komplementer kontinu, dan nilai absolut energi bebas kurang dari 4,5, sedangkan primer belakang menunjukkan kontinyu 6 basa komplementer, dan energi bebasnya 8,8;selain itu, dimer yang lebih serius muncul di ujung 3′, dan dimer dari 4 basa berurutan muncul.Meskipun energi bebasnya tidak tinggi, 3′ dimer Chl dapat secara serius memengaruhi spesifisitas amplifikasi dan efisiensi amplifikasi.Selain itu, perlu untuk memeriksa jepit rambut, heterodimer, dan ketidakcocokan.
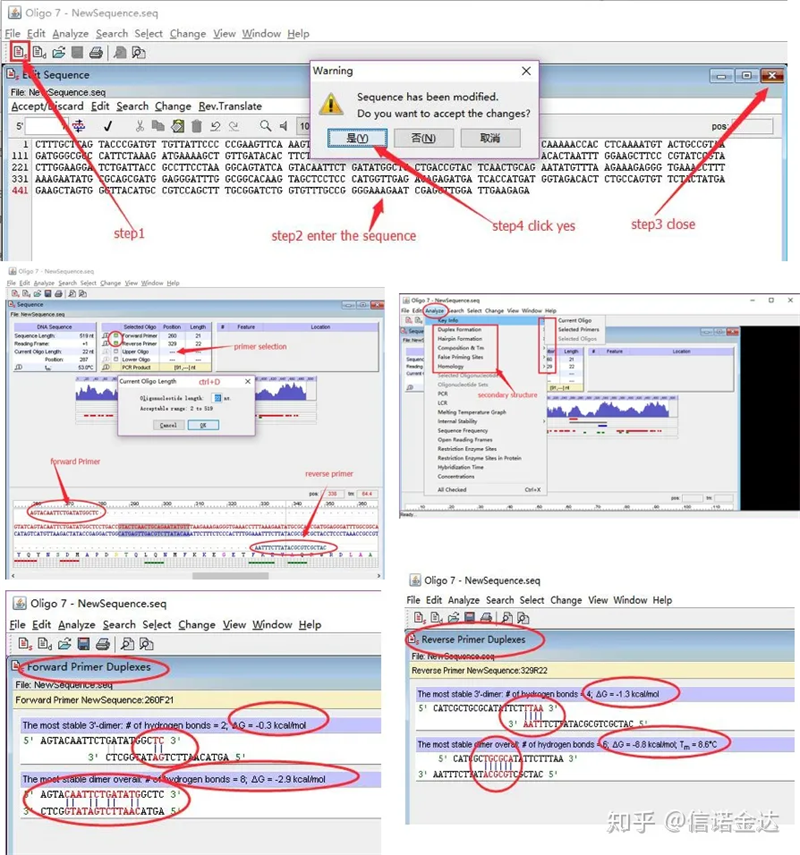
Hasil deteksi fig3 oligo7
Deteksi efisiensi amplifikasi:
Efisiensi amplifikasi reaksi PCR sangat mempengaruhi hasil PCR.Juga di qRT-PCR, efisiensi amplifikasi sangat penting untuk hasil kuantitatif.Hapus zat, mesin, dan protokol lain di buffer reaksi.Kualitas primer juga memiliki pengaruh besar pada efisiensi amplifikasi qRT-PCR.Untuk memastikan keakuratan hasil, kuantifikasi fluoresensi relatif dan kuantifikasi fluoresensi absolut perlu mendeteksi efisiensi amplifikasi primer.Diakui bahwa Efisiensi amplifikasi qRT-PCR yang efektif adalah antara 85% dan 115%.Ada dua metode:
1. Metode kurva standar:
A.Campurkan cDNA
B.Pengenceran gradien
c.qPCR
D.Persamaan regresi linier untuk menghitung efisiensi amplifikasi
2.LinRegPCR
LinRegPCR adalah program untuk analisis Data RT-PCR waktu nyata, juga disebut data PCR kuantitatif (qPCR) berdasarkan SYBR Green atau kimia serupa.Program ini menggunakan data yang dikoreksi non-baseline, melakukan koreksi baseline pada setiap sampel secara terpisah, menentukan window-of-linearity dan kemudian menggunakan analisis regresi linier agar sesuai dengan garis lurus melalui kumpulan data PCR.Dari kemiringan garis ini, efisiensi PCR dari masing-masing sampel dihitung.Rata-rata efisiensi PCR per amplikon dan nilai Ct per sampel digunakan untuk menghitung konsentrasi awal per sampel, yang dinyatakan dalam satuan fluoresensi arbitrer.Input dan output data melalui spreadsheet Excel.Hanya sampel
pencampuran diperlukan, tidak ada gradien
diperlukan langkah-langkah:(Ambil Bole CFX96 sebagai contoh, bukan Mesin dengan ABI yang jelas)
percobaan:itu adalah eksperimen qPCR standar.
keluaran data qPCR:LinRegPCR dapat mengenali dua bentuk file keluaran: RDML atau hasil Kuantifikasi Amplifikasi.Faktanya, ini adalah nilai deteksi waktu nyata dari nomor siklus dan sinyal fluoresensi oleh mesin, dan amplifikasi diperoleh dengan menganalisis nilai perubahan fluoresensi dari efisiensi segmen linier.
Pemilihan data: Secara teori, nilai RDML harus dapat digunakan.Diperkirakan masalah komputer saya adalah perangkat lunak tidak dapat mengenali RDML, jadi saya memiliki nilai keluaran excel sebagai data asli.Disarankan untuk melakukan penyaringan kasar data terlebih dahulu, seperti kegagalan menambahkan sampel, dll. Titik-titik tersebut dapat dihapus dalam data keluaran (tentu saja, Anda tidak dapat menghapusnya, LinRegPCR akan mengabaikan titik-titik ini di tahap selanjutnya)
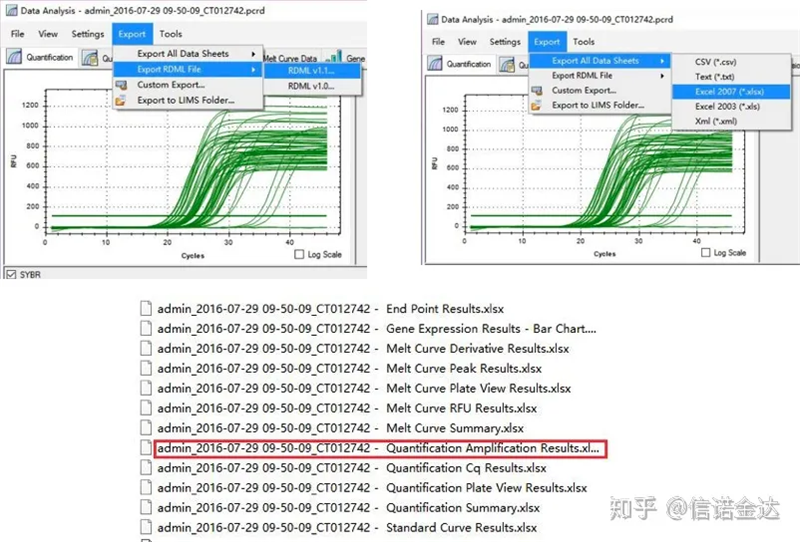
Fig5 ekspor data qPCR
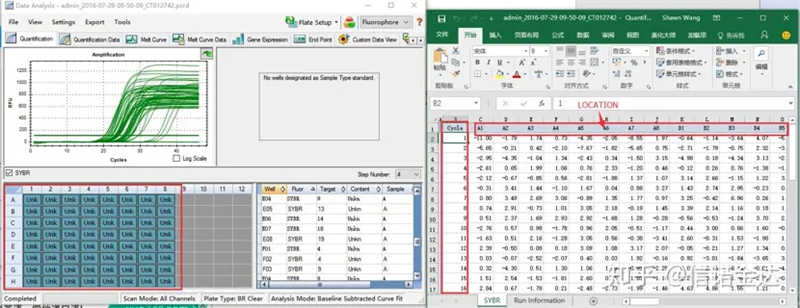
Gambar 6 pemilihan kandidat sampel
Masukan data:Buka hasil amplifikasi kualifikasi.xls, → buka LinRegPCR → file → baca dari excel → pilih parameter seperti pada Gambar 7 → OK → klik tentukan garis dasar
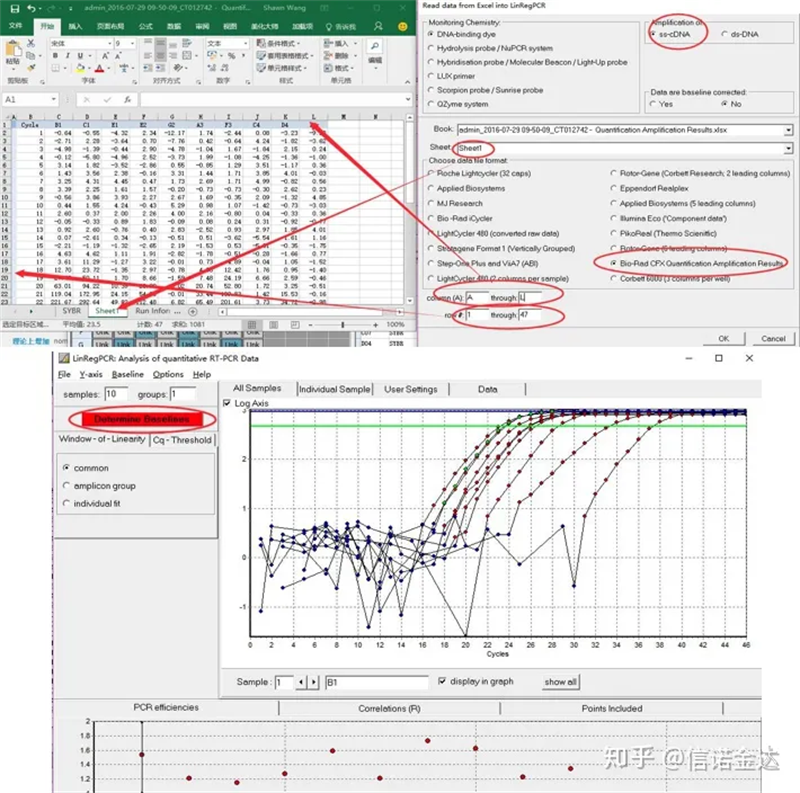
Fig7 langkah input data linRegPCR
Hasil:Jika tidak ada pengulangan, tidak diperlukan pengelompokan.Jika ada pengulangan, pengelompokan dapat diedit dalam pengelompokan sampel, dan nama gen dimasukkan dalam pengenal, dan kemudian gen yang sama akan dikelompokkan secara otomatis.Terakhir, klik file tersebut, ekspor excel, dan lihat hasilnya.Efisiensi amplifikasi dan hasil R2 dari setiap sumur akan ditampilkan.Kedua, jika Anda membagi menjadi beberapa grup, efisiensi amplifikasi rata-rata yang dikoreksi akan ditampilkan.Pastikan efisiensi amplifikasi setiap primer antara 85% dan 115%.Jika terlalu besar atau terlalu kecil, itu berarti efisiensi amplifikasi primer buruk.
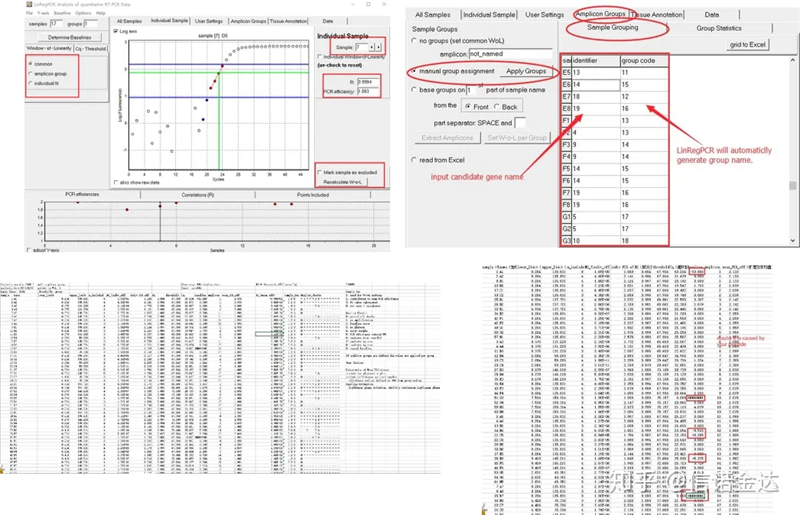
Gambar 8 Hasil dan keluaran data
Proses eksperimental:
Persyaratan kualitas RNA:
Kemurnian:1.72.0 menunjukkan bahwa mungkin ada sisa isotiosianat.Asam nukleat bersih A260/A230 harus sekitar 2 .Jika ada serapan kuat pada 230 nm, ini menunjukkan adanya senyawa organik seperti ion fenat.Selain itu, dapat dideteksi dengan elektroforesis gel agarosa 1,5%.Arahkan penanda, karena ssRNA tidak memiliki denaturasi dan logaritma berat molekul tidak memiliki hubungan linier, dan berat molekul tidak dapat dinyatakan dengan benar.Konsentrasi: Secara teoritisbukankurang dari 100ng/ul, jika konsentrasinya terlalu rendah, kemurniannya umumnya rendah tidak tinggi
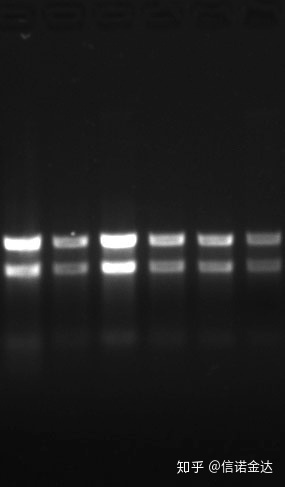
Gel RNA Fig9
Selain itu, jika sampel sangat berharga dan konsentrasi RNA tinggi, dianjurkan untuk memisahkannya setelah ekstraksi, dan mengencerkan RNA hingga konsentrasi akhir 100-300ng/ul untuk transkripsi balik.Di dalamproses transkripsi balik, ketika mRNA ditranskripsi, primer oligo (dt) yang secara spesifik dapat berikatan dengan ekor poliA digunakan untuk transkripsi balik, sedangkan lncRNA dan circRNA menggunakan primer hexamer acak (Random 6 mer) untuk transkripsi balik total RNA Untuk miRNA, primer loop leher khusus miRNA digunakan untuk transkripsi balik.Banyak perusahaan kini telah meluncurkan kit tailing khusus.Untuk metode loop batang, metode tailing lebih nyaman, throughput tinggi, dan hemat reagen, tetapi Efek membedakan miRNA dari keluarga yang sama seharusnya tidak sebaik metode loop batang.Setiap kit transkripsi balik memiliki persyaratan untuk konsentrasi primer spesifik gen (stem-loop).Referensi internal yang digunakan untuk miRNA adalah U6.Dalam proses pembalikan batang-loop, tabung U6 harus dibalik secara terpisah, dan primer depan dan belakang U6 harus ditambahkan secara langsung.Baik circRNA dan lncRNA dapat menggunakan HKG sebagai referensi internal.Di dalamdeteksi cDNA,
jika tidak ada masalah dengan RNA, cDNA juga akan baik-baik saja.Namun, jika kesempurnaan percobaan dikejar, yang terbaik adalah menggunakan gen referensi internal (Gen referensi, RG) yang dapat membedakan gDNA dari cd.Secara umum, RG adalah gen rumah tangga., HKG) seperti terlihat pada Gambar 10;Saat itu saya sedang membuat protein simpanan kedelai, dan menggunakan aktin7 yang mengandung intron sebagai referensi internal.Ukuran fragmen yang diamplifikasi dari primer ini dalam gDNA adalah 452bp, dan jika cDNA digunakan sebagai templat, ukurannya adalah 142bp.Kemudian hasil tes menemukan bahwa Bagian dari cDNA sebenarnya terkontaminasi oleh gDNA, dan itu juga membuktikan bahwa tidak ada masalah dengan hasil transkripsi balik, dan dapat digunakan sebagai template untuk PCR.Tidak ada gunanya menjalankan elektroforesis gel agarosa secara langsung dengan cDNA, dan ini adalah pita difus, yang tidak meyakinkan.
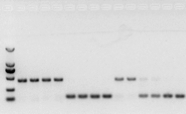
Gambar 10 deteksi cDNA
Penentuan kondisi qPCRumumnya tidak ada masalah menurut protokol kit, terutama pada langkah nilai tm.Jika beberapa primer tidak dirancang dengan baik selama desain primer, menghasilkan perbedaan besar antara nilai tm dan teori 60°C, direkomendasikan cDNA Setelah sampel dicampur, jalankan PCR gradien dengan primer, dan coba hindari pengaturan suhu tanpa pita sebagai nilai TM.
Analisis data
Metode pemrosesan PCR kuantitatif fluoresensi relatif konvensional pada dasarnya sesuai dengan 2-ΔΔCT.Templat pemrosesan data.
Produk-produk terkait:
PCR Waktu Nyata MudahTM –Taqman
PCR Waktu Nyata MudahTM –SYBR HIJAU I
RT Easy I (Master Premix untuk sintesis cDNA untai pertama)
RT Easy II (Master Premix untuk sintesis cDNA untai pertama untuk qPCR)
Waktu posting: Mar-14-2023



